

SCHEDULE
ABOUT US
本コンソーシアムでは、ナノテクノロジーの持続的発展を支える高度な実践力を有する人材を産学協同で育成するために、
1 大阪大学が推進する「ナノ高度学際教育研究訓練プログラム社会人教育」において、
- ・教育効果をより向上させるためのプログラム編成へのアドバイス
- ・受講生の授業料負担を軽減するための資金支援
2 企業が実践する日常のナノテクノロジーに関する研究開発に関して、
- ・教育の成果を具現化するための技術や情報の相談・交流の場の提供
- ・テクノロジーにかかわる「社会受容、標準化、リスクアセスメント」や、「産業・技術ロードマップ活用によるデザイン力養成」など、
特定テーマのセミナーによる産学の広範な連携・協力を支援するための活動を実施します。
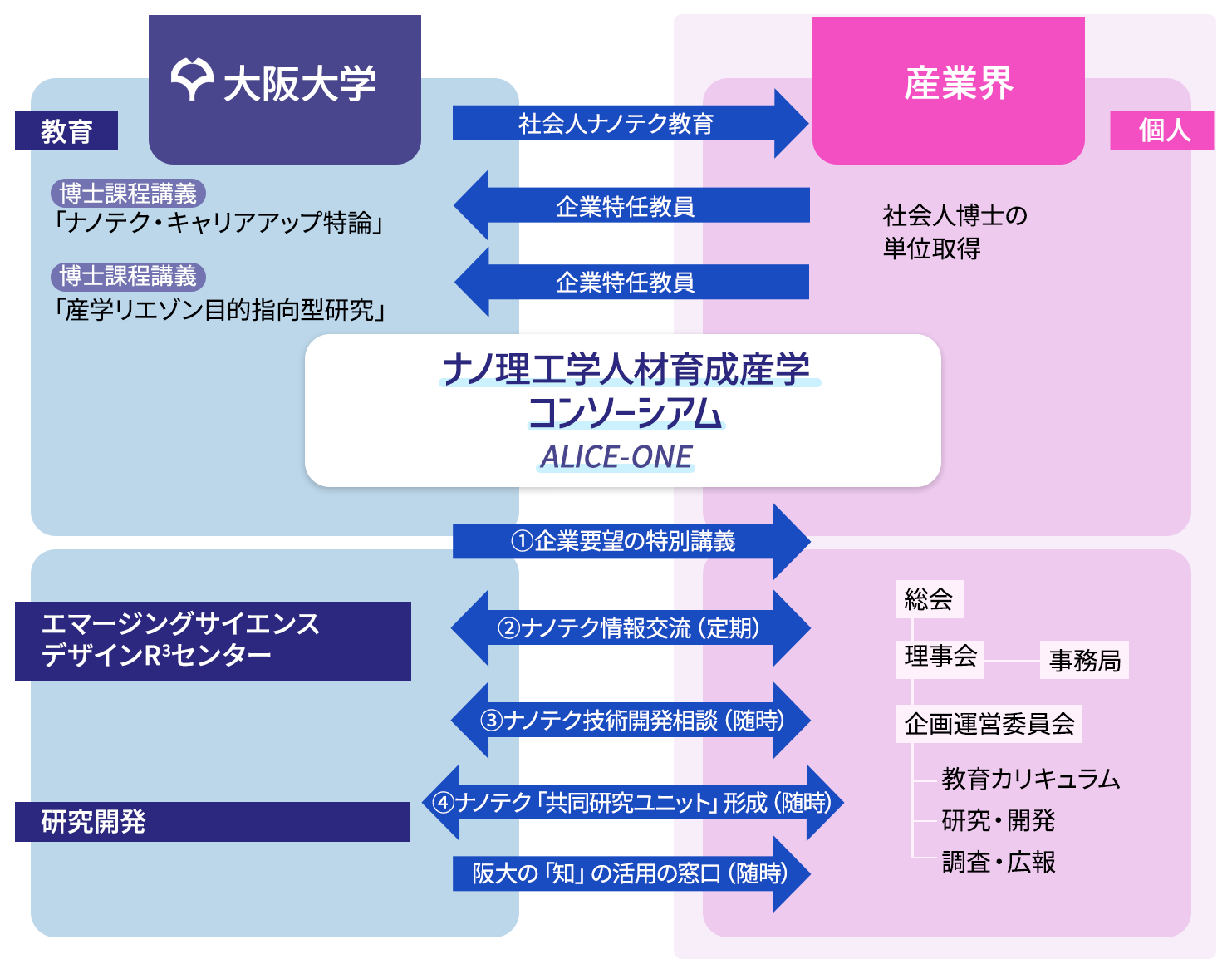
- ① 教育効果の一層の向上を図るため、化学力向上に必要な基礎学問に加えて産業界が求める有用な講義や実習を適切に配置したカリキュラムなどの検討
- ② 産学双方からのトピックス紹介(4回/年):討議4時間、会員以外 は有料
時々の関心あるテーマでのセミナー(1回/年):8時間、会員以外は有料 - ③ 単独もしくは複数の「会員企業と大学研究室」が特定のテーマで実施する技術開発の討議や共同開発を支援
- ④ 単独もしくは複数の「会員企業と大学研究室」が、研究開発推進の目的で学内に設置する共同研究ユニットを支援




